پیش بینی ساختار سه بعدی پروتئین (Prediction of Protins 3D Structure)
 مدل سازی همولوژیکی (homology modelling): اساس پیش بینی توسط مدل سازی همولوژیکی این است که توالی پروتئین با یک یا تعداد بیشتر پروتئین با ساختار شناخته شده شباهت داشته باشد. این روش، مدل سازی مقایسه ای (CA) نیز نامیده می شود بر اساس این واقعیت که پروتئین هایی با توالی ها ی مشابه ، ساختار های مشابه دارند . این روش بر آن اساس که ، پروتئین های یک خانواده، بیشترتوالی های آمینو اسیدی شان حفاظت شده است، آسان می باشد. چرا که وقتی ساختار یک پروتئین از یک خانواده به وسیله ی آزمایش تعیین شود، اعضای دیگر آن خانواده می توانند بر اساس تطابق شان با ساختار شناخته شده مدل سازی شوند. دقت پیش گویی با این روش به میزان تشابه بین توالی پروتئین هدف و ساختار های الگو بستگی دارد. مشکل عمده مدل سازی همولوژیکی پیدا کردن توالی هدفی است که به عنوان الگو استفاده می شود. تقریبا 57% همه ی توالی های شناخته شده حداقل یک دمین دارند که به یک پروتئین با ساختار شناخته شده وابسته می باشد.
مدل سازی همولوژیکی (homology modelling): اساس پیش بینی توسط مدل سازی همولوژیکی این است که توالی پروتئین با یک یا تعداد بیشتر پروتئین با ساختار شناخته شده شباهت داشته باشد. این روش، مدل سازی مقایسه ای (CA) نیز نامیده می شود بر اساس این واقعیت که پروتئین هایی با توالی ها ی مشابه ، ساختار های مشابه دارند . این روش بر آن اساس که ، پروتئین های یک خانواده، بیشترتوالی های آمینو اسیدی شان حفاظت شده است، آسان می باشد. چرا که وقتی ساختار یک پروتئین از یک خانواده به وسیله ی آزمایش تعیین شود، اعضای دیگر آن خانواده می توانند بر اساس تطابق شان با ساختار شناخته شده مدل سازی شوند. دقت پیش گویی با این روش به میزان تشابه بین توالی پروتئین هدف و ساختار های الگو بستگی دارد. مشکل عمده مدل سازی همولوژیکی پیدا کردن توالی هدفی است که به عنوان الگو استفاده می شود. تقریبا 57% همه ی توالی های شناخته شده حداقل یک دمین دارند که به یک پروتئین با ساختار شناخته شده وابسته می باشد. 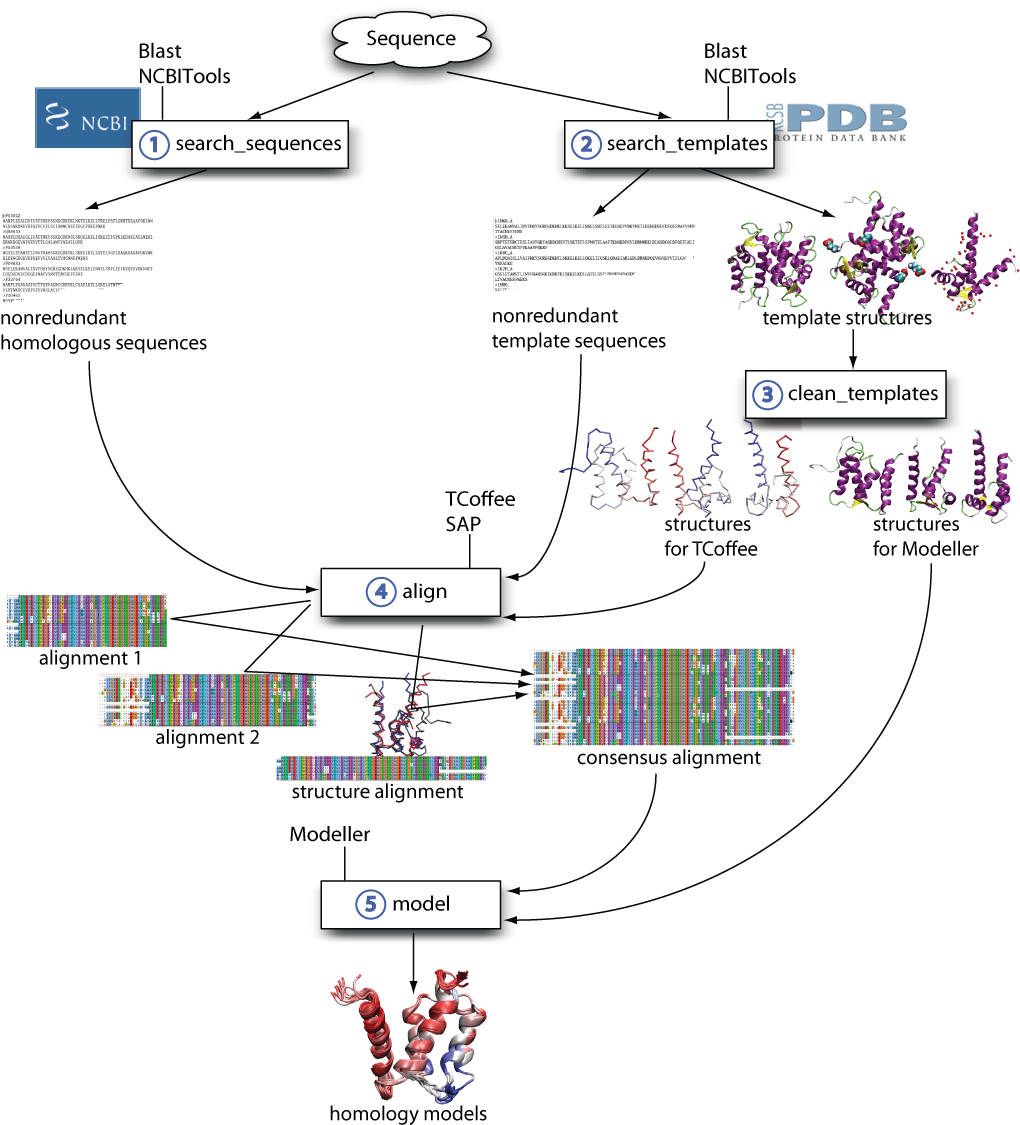 روش اب انیشیو (AB initio method)، همچنین روش دنو نامیده می شود. اصطلاح AB initio methodدر ابتدا اشاره به روش هایی داشت که برای پیش بینی ساختار پروتئین انجام می شد و از دانسته های آزمایشگاهی مربوط به ساختار استفاده نمیکرد. بعد ها این اصطلاح با به وجود آمدن روش هایی بر اساس فراگمنت ها مبهم شد، چرا که این روش ها براساس این واقعیت به کار می روند که گر چه ما نمی توانیم همه ی فولد ها ی استفاده شده در بیولوژی را ببینیم ، احتمالا قادر خواهیم بود تقریبا همه ی زیر ساختار ها را ببینیم . در این روش فرض می شود که کمترین انرژی آزاد در طول عمر پروتئین می بایست در دسترس باشد و تلاش می شود تا این مقدار کم را به وسیله بررسی بسیاری از کانفورماسیون های ممکن پروتئین به دست آورند. AB initio method به دو کلاس عمده تقسیم می شود : بندکشی پروتئین (threading ): که گاهی نیز تشخیص فولد (FR ) نامیده می شود.این روش یک رویکرد حد واسط دو روش قبلی است که هم از تشابه توالی در صورت وجود و هم از اطلاعات مربوط به تطابق های ساختاری استفاده می کند. هدف این روش تطابق توالی هدف با ساختار شناخته شده در کتابخانه ای از فولد ها می باشد .
روش اب انیشیو (AB initio method)، همچنین روش دنو نامیده می شود. اصطلاح AB initio methodدر ابتدا اشاره به روش هایی داشت که برای پیش بینی ساختار پروتئین انجام می شد و از دانسته های آزمایشگاهی مربوط به ساختار استفاده نمیکرد. بعد ها این اصطلاح با به وجود آمدن روش هایی بر اساس فراگمنت ها مبهم شد، چرا که این روش ها براساس این واقعیت به کار می روند که گر چه ما نمی توانیم همه ی فولد ها ی استفاده شده در بیولوژی را ببینیم ، احتمالا قادر خواهیم بود تقریبا همه ی زیر ساختار ها را ببینیم . در این روش فرض می شود که کمترین انرژی آزاد در طول عمر پروتئین می بایست در دسترس باشد و تلاش می شود تا این مقدار کم را به وسیله بررسی بسیاری از کانفورماسیون های ممکن پروتئین به دست آورند. AB initio method به دو کلاس عمده تقسیم می شود : بندکشی پروتئین (threading ): که گاهی نیز تشخیص فولد (FR ) نامیده می شود.این روش یک رویکرد حد واسط دو روش قبلی است که هم از تشابه توالی در صورت وجود و هم از اطلاعات مربوط به تطابق های ساختاری استفاده می کند. هدف این روش تطابق توالی هدف با ساختار شناخته شده در کتابخانه ای از فولد ها می باشد .روند کلی پیش بینی ساختار پروتئین در شکل زیر نشان داده شده است.
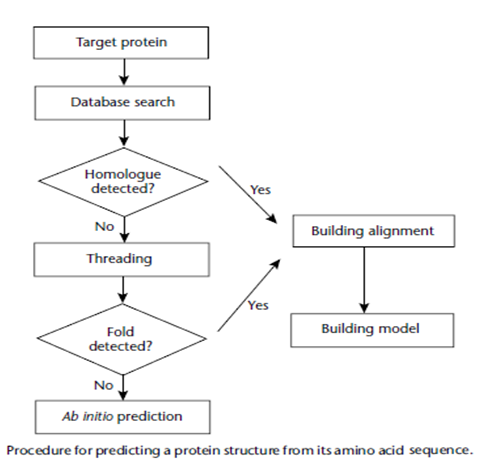
به دلیل تمایل زیاد پژوهشگران به پیش بینی ساختار پروتئین به وسیله ی روش های کامپیوتری ، جوامع علمی تلاشهای زیادی را در حل مشکلات مختلف و محدودیت های هر کدام از این روش ها دارند. که درنهایت پروتئین مورد نظر با بهترین ساختار استخراج گردد.

مرجع:
MIHĂŞAN M, Cuza AI: BASIC PROTEIN STRUCTURE PREDICTION FOR THE BIOLOGIST. 2010, 62(4):857 - 871
